Protein-Inorganic Material Interfaces
The primary goal of this research is to interrogate protein-inorganic material interface to study the biomineralization process of bones and teeth in the human body.
Molecular level investigation of biomolecular interfaces on bone:
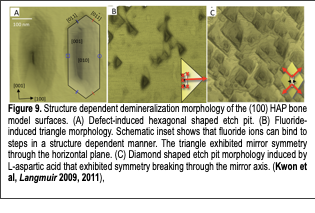
In our bodies, protein-inorganic crystal interfaces play a critical role in developing our bones and maintaining the mechanical integrity of the body. In order to interrogate protein-inorganic interfaces, we developed bone model surfaces whose structures were defined at the molecular level and investigated their structure-dependent characteristics using in situ atomic force microscopy techniques. Mature mammalian hard tissues are composed mainly of calcium phosphates in the form of mineralized biological apatites. Biological apatites vary in impurity content but are most closely related to hydroxyapatite (HAP), Ca10(PO4)6(OH)2. Tremendous efforts have been made to determine how various molecules interact with biologically derived and synthetic apatites in order to better understand their roles in hard tissue biomineralization and integrity. However, the interactions between bone crystal and other biological molecules are not understood at the molecular level due to the complexity of bone structures, the lack of a well-defined model system, and the lack of tools to investigate bone mineral’s complex interfaces. Therefore, we used two crystal surfaces [(100) and (001) HAP] as models to study events at the bone interface: The former was used to model the bone surface at which proteins interact, and the latter was used to model the surface at which bone crystal growth dominantly occurs, when interfaced with collagen. Using in situ atomic force microscopy techniques, we investigated these HAP crystals under precisely controlled fluid conditions and demonstrated the first detailed characterization of hydroxyapatite interface properties at the molecular level (Fig.9). Based on this crystal structure dependent (100) HAP model system, we investigated bone mineral interface properties under precisely controlled conditions and found multiple new discoveries related to bone and tooth health:
I. High ionic concentrations of NaCl suppress bone demineralization: Na+ and Cl- ions are the major contributors to the ionic concentration in our bodies. Our publication (Kwon et al, JPC 2009) showed that high ionic concentrations of NaCl play a critical role in stabilizing bone crystals. Our findings suggest that typical Na+ and Cl- ion concentrations (140 mM) in extracellular body fluids may play a significant role in bone mineral stability.
II. Bone defects accelerate the demineralization of bone crystal: Defects in bone minerals play a critical role in the bone remodeling processes. Our paper (Kwon et al, Langmuir, 2008) demonstrated that local structural defects on the model bone surfaces significantly accelerated demineralization (Fig. 9A). We characterized demineralization both in the presence and absence of defects. Our characterization showed that defects in bone could be easily removed by exposure to acidic buffers secreted by osteoclasts. Crystal surfaces with defects are dissolved ~100 times faster than non-defected surfaces.
III. The anti-cariogenic property of fluoride originates from the specific binding of fluoride to specific HAP steps: Fluoride ions play a critical role in preventing tooth decay. The work reported in our paper (Kwon et al, Langmuir 2011) verified the molecular mechanism behind the anti-cariogenic properties of fluoride. Through in situ dynamic characterization of steps on the (100) HAP surfaces under medically relevant fluoride concentrations (ranging from those found in tap water and mouth rinses), we measured the dynamic evolution of the crystal morphology. Changes to individual steps across a wide range of fluoride concentrations suggested that the anti-cariogenic property of fluoride ions originates from their strong interactions with the molecular steps in a structure dependent manner (Fig. 9B).
IV. Chiral interaction with L-aspartic acid induces chiral dissolution of bone surfaces: Organic modifiers (i.e., amino acids, peptides, and proteins) play critical roles in bone remodeling. Our work (Kwon et al, in preparation) presented the first detailed experimental investigation of specific structure dependent chiral interactions between L-aspartic acid (Asp) and HAP crystal surfaces (Fig. 9C). Aspartic acid residues are enriched in bone-associated proteins. Bone mineralization and demineralization are believed to be highly influenced by aspartic acid. We verified specific aspartic acid interactions with individual HAP molecular steps during the HAP dissolution process. Our study provides fundamental insight into the interactions between organic modifiers and bone crystals and expands our understanding of the morphogenesis of bone and tooth crystals.
Discovery of collagen-like peptide that nucleated HAP crystals:
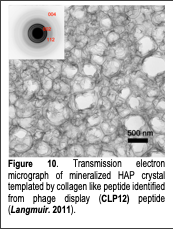
Based on our understanding of the bone model surfaces, we investigated the molecular level interactions between well-defined model bone surfaces (HAP (100) surfaces) and HAP-binding peptides discovered through phage display. The formation of natural bone is thought to occur by the templated mineralization of HAP by surrounding proteins, which include collagen and highly acidic phosphoproteins associated with the collagen scaffold. It has been proposed that the acidic groups serve as binding sites for calcium ions and align them in an orientation that matches the HAP crystal lattice. However, the biological mineralization process is not understood at the molecular level. An appealing strategy for biomimetic bone synthesis would be to employ HAP-binding peptides to facilitate HAP crystal growth. Unfortunately, the rational design of such peptides is deterred by a lack of knowledge regarding the sequences that guide HAP mineralization in vivo.
Using phage display, we identified a 12-residue peptide that bound to single crystal (100) HAP surfaces under physiological pH conditions (pH 7.5) (Langmuir 2011). This peptide was able to template the nucleation and growth of crystalline HAP minerals in a sequence- and composition-dependent manner (Fig. 10). The sequence responsible for the mineralizing activity resembled the tripeptide repeat (Gly-Pro-Hyp; Hyp: Hydroxyproline) of type I collagen, a major component of the bone extracellular matrix. Using a panel of synthetic peptides, we defined the structural features required for mineralizing activity. The results suggest a model for the cooperative non-covalent interaction of the peptide with HAP and support the theory that native collagen has a mineral-templating function in vivo.
Microscopic level interactions at bone biointerfaces
We investigated microscopic bone protein-crystal interactions to develop biomimetic, organic-inorganic nanocomposite materials. The organic component of bone materials is comprised of collagenous and non-collagenous protein matrices. Hierarchical collagenous protein matrices are known to provide a structural framework for templating mineralized bone crystals, whereas non-collagenous proteins in bone bind the mineral phase, transfer strain, and dissipate energy through the breaking of ‘sacrificial bonds’. These proteins form various bonds through non-covalent intra-chain, inter-chain, and interfacial interactions. Because of their importance in nature, it would be extremely beneficial to further explore the rational design of synthetic organic phase analogues. Because conventional approaches of utilizing off-the-shelf polymers and naturally derived biological materials are very limited and fail to emulate the highly tuned sequences of natural proteins, we designed sequence specific biomacromolecules that closely emulate the function of bone protein matrices using biological approaches. Specifically, we developed sequence specific elastin-based protein matrices and phage-based collagen-mimetic nanofiber matrices.
Bioinspired elastin-based bone nanocomposite materials:
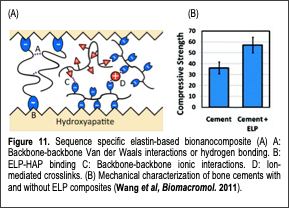
Non-collagenous protein matrices play multiple critical roles in bone mechanical properties. Our paper (Wang et al, Biomacromolecules 2011) presented our approach of emulating the mechanical enhancements observed in nature by designing sequence specific protein-based polymers and incorporating them into calcium phosphate bone cement materials. We developed novel elastin-like polymers (ELP) to construct mechanically enhanced calcium phosphate cement (CPC)-protein nanocomposites. By controlling ELP primary structures at the molecular level through genetic engineering, we could synthesize protein matrices to mimic the adhesive proteins of bone. We incorporated octaglutamic acid, a previously characterized HAP binding motif, at the protein termini and positively, negatively, or neutrally charged amino acids along the polymer backbone to control intramolecular, intermolecular, and interfacial interactions with HAP. By imparting ELPs with varying modes of inter/intra molecular bonding and interfacial bonding, we were able to demonstrate sequence specific property changes in ELP-HAP mixtures (Fig. 11A). Our results showed that interfacial binding to HAP through octaglutamic acid motifs was critical for imparting functionalities such as nanoparticle dispersion and mechanical property improvements in ELP-CPC composites (Fig. 11B). In the absence of binding, the physical properties of the protein had no effect. We also demonstrated the injectability of the ELP-CPC composites, which transformed into nanocomposites at body temperature in vitro. We believe that our results are not limited to applications involving HAP but can be expanded to explore composites containing other metals and minerals. Our design and synthesis approaches may yield strong and tough materials for future load-bearing applications such as hard-tissue replacements.
Light responsive hydrogel elastin-graphene nanocomposite materials:
We expanded our elastin-based composite material design theme to develop a bio-inspired light-responsive soft-robotic materials using protein engineering. Hydrogel actuators (HAs) are water swollen polymer networks that reversibly change their dimensions or shapes when external stimuli (e.g., solvent composition, temperature, and external fields) cause local or global changes in network swelling. HAs with tunable speed and motion are highly sought after to fulfill applications in various fields, including biology, medicine, microfluidics, and robotics. To fulfill the bio-related applications, HAs which utilize materials and stimuli that are compatible with biomolecules and cells are also desired.
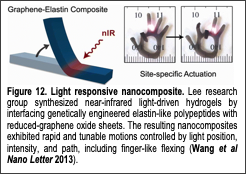
Light-driven HAs are particularly attractive systems but have limitations that must be addressed to expand their utility. Light is applied remotely, so solution-wide changes and the introduction of potentially invasive wires or electrodes are avoided. In addition, light is easily controlled with higher spatial and temporal resolution than other stimuli. However, macroscale, photothermal HAs exhibit slow actuation kinetics (on the order of minutes), due to limited rates of water diffusion into and out of the networks. In addition, previous photothermal HAs had isotropic structures that changed size relatively evenly in each dimension, making non-linear motions difficult to achieve. In contrast, actuation into more complex shapes can be achieved by creating hydrogel networks with anisotropic composition, crosslinking, or porosity. With these limitations in mind and with an eye towards future bio-related applications, we created light-driven HAs that could rapidly undergo non-linear motions by combining reduced graphene oxide (rGO) nanosheets and elastin-like polypeptides (ELPs).
In our recent work (Wang et al, Nano Letters 2013) we synthesized a new chimeric ELP fused with the graphene binding (GB) peptide ‘HNWYHWWPH’. This ELP-GB can physically bind to reduced graphene oxide (rGO) and graphene oxide (GO) via π-π stacking and keep the nanomaterials dispersed. The composite created as a result is light-responsive as rGO and GO act as photothermal heaters by absorbing light and locally generating heat while ELP absorbs the heat and responds mechanically by contracting. The ELP-GB chimera was further modified with the addition of an/the ‘RGD’ cell adhesive peptide (Wang et al, Langmuir 2014) to show the ease of improving cell behavior on these composites. We also synthesized hydrogels using the ELP-GB composites along with an additional ELP containing a lysine residue near the C-terminus; the primary amine groups were cross-linked together using an N-hydroxysuccinimide functional 4-armed PEG linker in organic solvents. A simple method of creating anisotropic gels was employed by allowing water vapor to freely diffuse into the water-miscible organic solvents as the gel cross-linked and the resulting gel contained distinct porous and solid layers. This gel is near infrared (nIR) light responsive as the rGO absorbs nIR and generates heat that causes ELP to transition and the gel to contract. Instead of an isotropic contraction, the porous layer of the gel contracts much faster than the solid side, resulting in bending of the gel. We demonstrated precise spatial and temporal control of the light-responsive actuation of these hydrogels (Fig. 12) raising the potential of these soft actuators for application as light-responsive drug delivery devices as well as remotely controllable soft actuators.
Self-sealing Biomedical Adhesive:
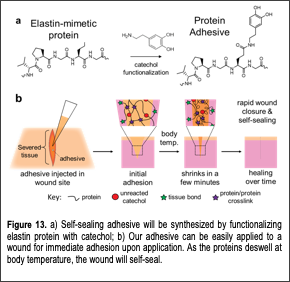
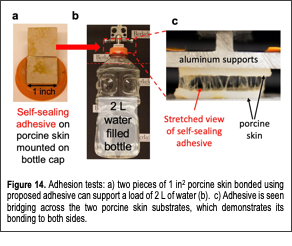
We developed a novel class of protein-based self-sealing medical adhesives (Desai et al, Biomacromolecules, 2020). Medical adhesives are important for reconnecting Text Box: Figure 13. a( Self-sealing adhesive will be synthesized by functionalizing elastin protein with catechol; b( Our adhesive can be easily applied to a wound for immediate adhesion upon application. As the proteins deswell at body temperature, the wound will self-seal. tissues during surgeries and wound repair. Typically, severed tissues are bonded using sutures and staples, which are effective, but cause further trauma by puncturing the target tissues. Although suturing is common, achieving the best functional and aesthetic outcome requires crucial time during surgery and high skill for the surgeon to achieve layer-by-layer tissue approximation. Simple-to-apply adhesives can eliminate the complications; however, currently available products have found poor adoption among surgeons due to unreliable performance. To create a robust adhesive, we developed engineered elastic proteins fused with mussel-inspired adhesive molecule, catechol (Fig. 13A). Our elastic proteins are synthetic versions of mammalian elastin that give high durability to tissues like blood vessels. In addition to flexibility, the elastin-mimetic proteins can shrink at body temperature (Fig. 13B) and impart robustness to the material unlike most hydrogels that undergo high swelling. Functionalization of the proteins with mussel inspired adhesive molecules (catechol) allows them to bond to biological tissues. The elastic protein-based adhesives make for a formidable adhesive as they can form strong bonds and self-seal the wound site by shrinking at body temperature. Compared to sutures, our engineered protein-based adhesives (Fig. 14) can drastically simplify layer-by-layer tissue approximation during wound closure. This is because an adhesive can be applied to all layers, which can then be aligned in a single step. This is in addition to their ability to stop bleeds. However, conventional cyanoacrylate-based glues are only FDA approved for topical use due to high toxicity; whereas, biocompatible alternatives such as hydrogels are prone to swelling that lead to further softening of a material that is already soft. Our proposed adhesives possess multiple advantages over conventional products. 1) Thermo-responsiveness of elastin-mimetic proteins will allow for self-sealing of wounds at body temperature. 2) Hydrophobic nature of the adhesive also ensures that it will not swell. 3) Through rational protein design, adhesive hydrogels composed of elastin-mimetic proteins can be ultra-flexible. 4) Elastin-mimetic proteins are biocompatible and genetic engineering can be used to customize the proteins by adding peptide motifs for tissue-specific cell signaling, enzymatic degradation and biomineralization. 5) Since the elastin-mimetic proteins are recombinant proteins, there Text Box: Figure 14. Adhesion tests: a( two pieces of 1 in2 porcine skin bonded using proposed adhesive can support a load of 2 L of water (b). c( Adhesive is seen bridging across the two porcine skin substrates, which demonstrates its bonding to both sides. is no batch-to-batch variability in protein size and composition. Achieving consistent material properties and function will make material synthesis scalable in the future.
We strongly believe that many surgeons and patients will benefit from our innovation. Our adhesives will be ideal for surgeries that require proper tissue approximation. These include anastomoses during gastrointestinal surgeries and wound closure by plastic surgeons to prevent scarring. In addition, the non-absorbent properties of the adhesive make it applicable for preventing seromas in plastic surgery, which involve large surface area wounds, such as breast reconstruction and abdominoplasties, and in orthopedic interventions, which involve post-surgery joint swelling, such as arthroscopies and joint replacements. The adhesives will also enable faster recovery from injuries for civilians and soldiers.
Elastin-Based Hard Tissue Engineering Materials:
We developed elastin-based hard tissue engineering materials through the collaboration with Prof. Sun-Young Kim (a former visiting scholar in our lab and a professor in School of Dentistry, Seoul National University; Jang et al, Scientific Report, 2018 & Journal of Biomedical Materials, 2020). Calcium phosphate cements (CPCs), which are usually composed of various calcium phosphate powder mixtures and a liquid solution, have been widely used as synthetic bioactive hard tissue engineering materials. The setting reaction is initiated by hydrolysis between the components, followed by dissolution and reprecipitation. Due to their compositional similarities to hydroxyapatite (HA), CPCs are valued as self-setting, bioactive, osteoconductive, osteotransductive, mouldable or injectable materials. These characteristics make CPCs promising materials for hard tissue engineering such as bone repair, replacement, and regeneration. Another beneficial feature of CPCs is their microporous structure, which makes them useful drug delivery vehicle for antibiotics, anti-inflammatory drugs, and antitumor drugs. They are also used to develop dental materials for the augmentation or repair of alveolar bone, tooth replacement, root surface desensitization, and root canal fillings. An ideal material for hard tissue engineering should have good mechanical strength to withstand loads, appropriate setting time, and favorable anti-washout capability to maintain stability in biological fluids. Despite their biocompatibility and easy applicability, the poor mechanical properties of CPCs limit their application at load-bearing sites. Moreover, their low cohesion means that CPCs are easily disrupted on premature contact with biological fluids or blood, leading to undesired washout. To overcome these limitations, we developed a novel composites to combine CPC and genetically engineered elastin-like polypeptides (ELPs). We investigated the effect of the ELPs on the physical properties and biocompatibility of CPC by testing ELP/CPC composites with various liquid/powder ratios in vitro and in vivo. Our results show that the addition of ELPs improved the mechanical properties of the CPC, including the microhardness, compressive strength, and washout resistance. The biocompatibility of ELP/CPC composites was also comparable to that of the CPC alone. Supplementing CPC with ELPs functionalized with octaglutamate as a hydroxyapatite binding peptide increased the setting time of the cement. With further design and modification of our biomolecules and composites, our elastin-based composite materials will lead to products with diverse applications hard tissue regeneration.